

Developer
COMPACT is a framework for performing CO-expression Module Perturbation Analysis in Cellular Transcriptomes. Building on co-expression networks from hdWGCNA, COMPACT applies direct perturbations to network hub genes and propagates perturbation signals through linked genes in the network. The package supports knock-in, knock-down, and knock-out perturbation strategies across different networks, gene sets, and cell lineages.
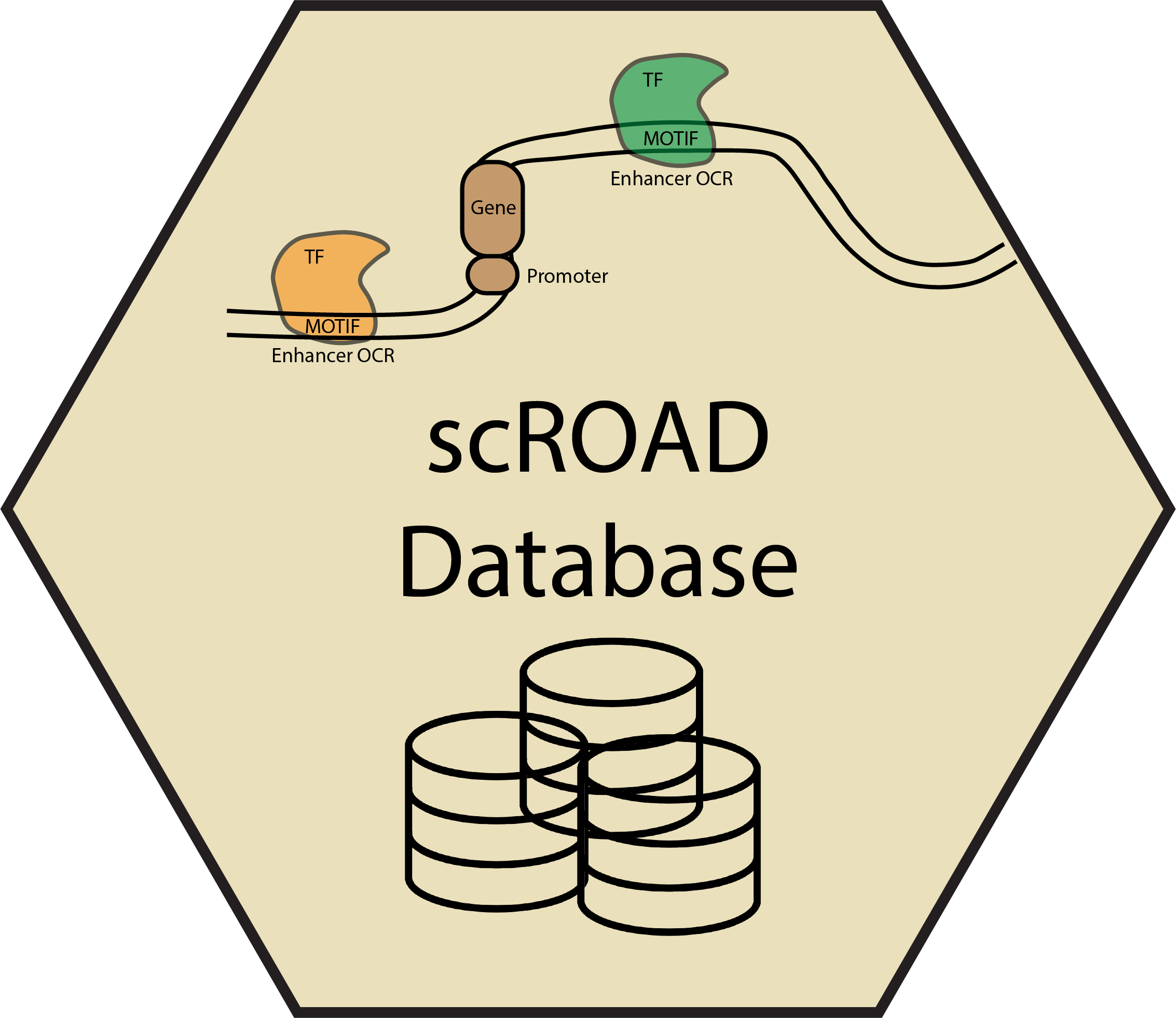
scROAD database offers comprehensive information on single-cell cCRE transcription factor occupancy data generated from snATAC-seq analysis of human postmortem prefrontal cortex (PFC) tissue. The data specifically focuses on Alzheimer's Disease and Pick's Disease. I developed this interactive database, scROAD, which integrates single-cell chromatin data processed with Signac, cis-regulatory links inferred by Cicero, and TF binding occupancy profiles generated using TOBIAS. We performed single cell co-accessibility analyses using Cicero to construct putative cis-regulatory enhancer-promoter links. Additionally, with the help from TOBIAS package, we can further explore transcription factor (TF) binding occupancy in ATAC-seq. This analysis allows us to detect differences in TF binding between disease and control samples, providing insights into how regulatory mechanisms are altered in specific cell types. By integrating TF binding data with co-accessibility analyses to create this scROAD interactive database, users can easily explore transcription factor binding activity and their implications in disease, providing a valuable resource for understanding gene regulation in neurodegeneration. For a more in-depth understanding of the database's purpose and contents, please refer to the Science Advances paper.


ArchRtoSignac is an R package to convert an ArchRProject [ArchR] to a Signac SeuratObject [Signac]. ArchR and Signac are both commonly used scATAC-seq analysis packages with comparable sets of features and are currently under development, which means they are likely to change over time. You can choose to use only one of these packages; however, you may want to use both packages for your analysis. For example, we use ArchR to generate a fixed-width peak matrix due to its computational advantage, and we use Signac for reference mapping to assist in cell-type annotation. Here we provide an option to help with the data formatting from an ArchRProject to a Signac SeuratObject: ArchRtoSignac, a wrapper function that allows easier implementation of both pipelines. In addition, conversion to a SeuratObject allows the use of other packages available through SeuratWrappers.
Contributor

hdWGCNA is an R package for performing weighted gene co-expression network analysis [WGCNA] in high dimensional transcriptomics data such as single-cell RNA-seq or spatial transcriptomics. hdWGCNA is highly modular and can construct context-specific co-expression networks across cellular and spatial hierarchies. hdWGNCA identifies modules of highly co-expressed genes and provides context for these modules via statistical testing and biological knowledge sources. hdWGCNA uses datasets formatted as [Seurat] objects.